Basic concepts¶
In the first chapter we will elaborate on how Nextflow is designed, its advantages and disadvantages, the basic components, etc.
Introduction¶
Writing workflows to automate processes is not something new. In the data/ folder we’ve written a bash script that downloads the data that we will use throughout this tutorial. These bash scripts are probably one of the oldest forms of workflows. Let’s have a look at another example:
#!/bin/bash
blastp -query sample.fasta -outfmt 6 \
| head -n 10 \
| cut -f 2 \
| blastdbcmd -entry - > sequences.txt
Starting with a shebang line, the blastp command is piped through multiple times to eventually result in an output file sequences.txt. The downside of this very basic and intuitive pipeline is that it has a sequential flow. In response to that, pipeline tools were built which are aimed to deal with more complex situations. Nextflow is designed around the idea that Linux has many simple but powerful command-line and scripting tools that, when chained together, facilitate complex data manipulations.
By definition, Nextflow is a reactive workflow framework and a programming Domain Specific Language that eases the writing of data-intensive computational pipelines[1]. Nextflow scripting is an extension of the Groovy programming language, which in turn is a super-set of the Java programming language. Groovy can be considered as Python for Java in a way that simplifies the writing of code and is more approachable.

Why (not)?¶
Nextflow is not the only player in the field[2], however there are good reasons to opt for it.
Parallelization: processes are automatically scheduled based on available resources
Scalability: simple scaling from local to HPC-cluster usage
Portability: run across different platforms
Reproducible: native support for containers, conda environments, and interaction with Git.
Continuous checkpoints for resuming / expanding pipelines (which is usually the case for workflow pipelines)
Community[3]
Some thoughts or disadvantages from my personal point of view, it takes some time to get used to the syntax of the Groovy language. As flexible as it is, as complex it gets. Often it’s difficult to trace down the exact problem of a failure of a pipeline script, especially in the beginning. It’s probably not the first thing you should be concerned of if you’re doing a one-time analysis.
Main abstractions¶
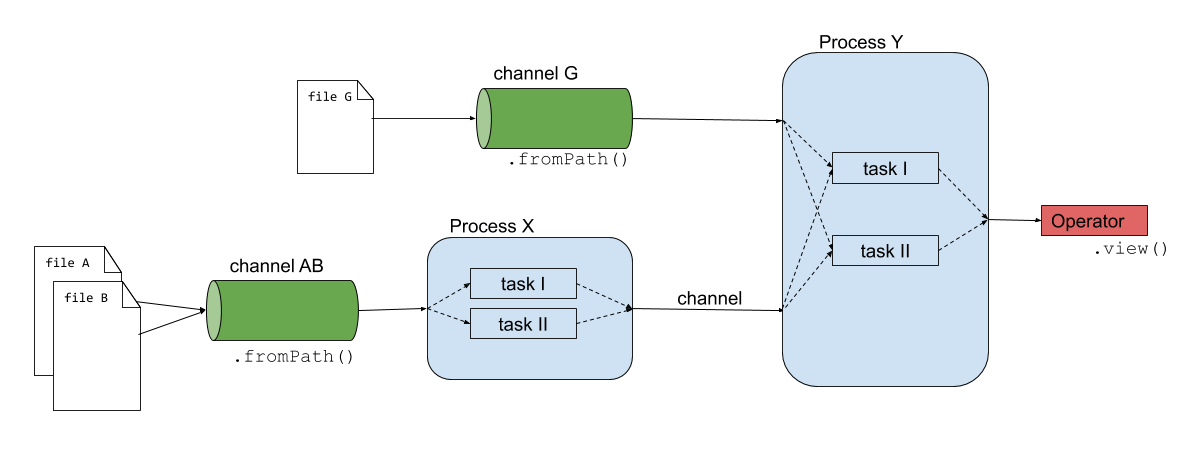
Nextflow consists of three main components: channels, operators and processes.
Channels: connect processes/operators with each other. On a more technical level, channels are unidirectional async queues that allows the processes to communicate with each other.
Operators: transform the content of channels by applying functions or transformations. Usually operators are applied on channels to get the input of a process in the right format.
Processes: define the piece of script that is actually being run (e.g. an alignment process with STAR)
The script02-basic-concepts/firstscript.nfis using these three components and gives an idea of how Nextflow scripts are being build.
Since the introduction of the new DSL2, workflows can be added to this list. This will be discussed in the next chapter.
1. Channels:¶
The input of the analysis is stored in a channel, these are generally files like sequencing, reference fasta, annotation files, etc. however the input can be of any kind like numbers, strings, lists, etc. To have a complete overview, we refer to the official documentation[4]. Here are some examples of how a channel is being created:
# Channel consisting of strings
strings_ch = Channel.from('This', 'is', 'a', 'channel')
# Channel consisting of a single file
file_ch = Channel.fromPath('data/sequencefile.fastq')
# Channel consisting of multiple files by using a wildcard *
multfiles_ch = Channel.fromPath('data/*.fastq')
# Channel consisting of multiple paired-end files by using wildcard * and options {x,y}
paired_ch = Channel.fromFilePairs('data/*{1,2}.fastq')
These channels can then be used by operators or serve as an input for the processes.
2. Operators:¶
Operators are necessary to transfor the content of channels in a format that is necessary for usage in the processes. There is a plethora of different operators[5], however only a handful are used extensively. Here are some examples that you might come accross:
collect: e.g. when using a channel consisting of multiple independent files (e.g. fastq-files) and need to be assembled for a next process.
Channel
.from( 1, 2, 3, 4 )
.collect()
.view()
# outputs
[1,2,3,4]
mix: e.g. when assembling items from multiple channels into one channel for a next process (e.g. multiqc)
c1 = Channel.from( 1,2,3 )
c2 = Channel.from( 'a','b' )
c3 = Channel.from( 'z' )
c1 .mix(c2,c3)
# outputs
1
2
3
'a'
'b'
'z'
3. Processes:¶
Processes are the backbone of the pipeline. They represent each individual subpart of the analysis. In the code-snippet below, you can see that it consists of a couple of blocks: directives, input, output, when clause and the script.
process < name > {
[ directives ]
input:
< process inputs >
output:
< process outputs >
when:
< condition >
[script|shell|exec]:
< user script to be executed >
}
Each process is executed independently and isolated from any other process. They communicate via asynchronous FIFO queues, i.e. one process will wait for the output of another and then runs reactively when the channel has contents.
Running our first pipeline:¶
If we want to run a Nextflow script in its most basic form, we will use the following command:
nextflow run example.nf
In our case, we will replace example.nf with 02-basic-consepts/firstscript.nf. First, inspect the script 02-basic-consepts/firstscript.nf and notice how the channels are being created, passed on to the process’ inputs, processed by the script section and then given to the output.
When we run this script, the result file will not be present in our folder structure. Where will the output of this script be stored?
Nextflow will generate an output that has a standard lay-out:
N E X T F L O W ~ version 20.07.1
Launching `02-basic-concepts/firstscript.nf` [elegant_curie] - revision: 9f886cc00a
executor > local (2)
executor > local (2)
[5e/195314] process > valuesToFile (2) [100%] 2 of 2 ✔
results file: /path/to/work/51/7023ee62af2cb4fdd9ef654265506a/result.txt
results file: /path/to/work/5e/195314955591a705e5af3c3ed0bd5a/result.txt
The output consists of:
Version of nextflow
Information regarding the script that has ran with an identifier name
Hash with process ID, progress and caching information
Optional output printed to the screen as defined in the script (if present) The results are stored in the results file as described in the two last lines. By default the results of a process are stored in the
work/directory in subfolders with names defined by the hashes.
Besides the output, also a bunch of hidden .command.* files are present:
.exitcode, contains 0 if everything is ok, another value if there was a problem.
.command.log, contains the log of the command execution. Often is identical to .command.out
.command.out, contains the standard output of the command execution
.command.err, contains the standard error of the command execution
.command.begin, contains what has to be executed before .command.sh
.command.sh, contains the block of code indicated in the process
.command.run, contains the code made by nextflow for the execution of .command.sh and contains environmental variables, eventual invocations of linux containers etc
Earlier, we described that Nextflow uses an asynchronous FIFO principle. Let’s exemplify this by running the script 02-basic-consepts/fifo.nf and inspect the order that the channels are being processed.
N E X T F L O W ~ version 20.07.1
Launching `02-basic-concepts/fifo.nf` [nauseous_mahavira] - revision: a71d904cf6
[- ] process > whosfirst -
This is job number 6
This is job number 3
This is job number 7
This is job number 8
This is job number 5
This is job number 4
This is job number 1
This is job number 2
This is job number 9
executor > local (10)
[4b/aff57f] process > whosfirst (10) [100%] 10 of 10
A script, as part of the process, can be written in any language (bash, Python, Perl, Ruby, etc.). This allows to add self-written scripts in the pipeline. The script can be written in the process itself, or can be present as a script in another folder and is run from the process here.
#!/usr/bin/env nextflow
process python {
"""
#!/usr/bin/python3
firstWord = 'hello'
secondWord = 'folks'
print(f'{firstWord} {secondWord}')
"""
}
Note
Directives will be handled further on in the course, conditionals are not considered here.